Ukupno je 45 različitih bolesti lizosomalnog skladištenja, koji su heterogena skupina urođenih metaboličkih bolesti. Ljudi koji pate od bilo koje od ovih bolesti imaju genetski defekt. Sve bolesti skladištenja imaju jedno zajedničko: određeni enzim je odsutan ili je djelomično funkcionalan.
Što je lizosomalna bolest skladištenja?

© designua - stock.adobe.com
Te su urođene bolesti skladištenja rijetke jer ih boluje manje od pet na 10 000 ljudi. Razne bolesti imaju vrlo različit tijek, a simptomi mogu jako varirati.
Najpoznatiji oblici bolest lizosomalnog skladištenja su Fabryjeva bolest, Gaucherova bolest, Pompeova bolest i mukopolisaharidoza (MPS). Često ih nazivaju "siročad lijekovima", jer put do točno određene dijagnoze i odgovarajuće terapije može biti vrlo dug. Ponekad mogu proći godine kako bi oni koji su pogođeni otkrili što im se događa.
uzroci
Bolesti lizosomalnog skladištenja karakteriziraju određene oblike nasljednih metaboličkih bolesti. Pacijentima nedostaje važan enzim koji osigurava neometani rad metabolizma. U manje izraženom obliku ovaj enzim barem nije prisutan u dovoljnim količinama.
Zadaća enzima je da odlažu zagađivače i otpadne tvari koje se u ljudskom organizmu nakupljaju putem metabolizma preko lizosoma, ili da ih ponovno obrađuju na takav način da se simptomi ne pojave.
Ako postoji nedostatak enzima, ovaj nesmetano ciklus odlaganja više nije zajamčen. Štetne tvari naseljavaju se u stanicama i narušavaju metabolički ciklus. U početnoj fazi poremećaji nemaju vidljiv učinak, postoje samo nekoliko ograničenja. Međutim, ako ovaj metabolički poremećaj ostane neliječen kao rezultat nedostatka enzima, simptomi se umnožavaju jer stanice postaju vrlo proširene.
Simptomi, tegobe i znakovi
U najgorem slučaju ovi će propasti. Posljedice su oštećenja kostiju, živčanog sustava, slezene, bubrega, mišića ili srca. Fabryjeva bolest uzrokuje skladištenje masti (globotriaosylceramide, Gb3) u stanicama zbog smanjene ili odsutne enzimske aktivnosti. Ove neželjene naslage mogu dovesti do jake boli u nožnim prstima ili prstima, moždanog udara i oštećenja bubrega.
Dijagnoza i tijek bolesti
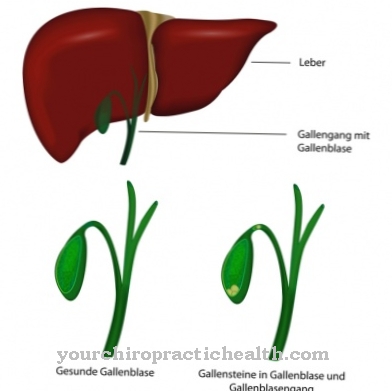
Ova klinička slika utječe na različite sustave istovremeno: krvne žile, bubrezi, srce i živčani sustav. Autosomno recesivna Gaucherova bolest izaziva mutaciju enzima "beta-glukocerebrosidaza" i dovodi do nakupljanja supstrata unutar stanica, posebno u makrofazima (fagocitima) koji pripadaju retikulo-endotelnom sustavu. Krvna slika se mijenja, jetra i slezina su povećane, a kosti boli.
Bolest je progresivna i uglavnom je etnička, jer se u većini slučajeva javlja kod ljudi židovskog porijekla. Pompeova bolest poznata je i kao "nedostatak kisele maltaze". Klinička slika pripada skupini glikogeneze tipa II. Pogođenim osobama nedostaje enzim "alfa-1,4-glukozidaza" (kisela maltaza) ili nije dostupan u dovoljnim količinama. Zbog poremećenog propadanja glikogena u mišićima, pacijenti pate od uništavanja mišićnih stanica u obliku skladištenja šećera.
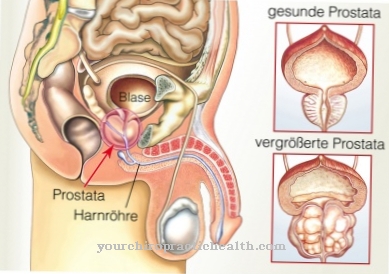
Mukopolisaharidoza tipa I (MPS), poznata i kao Hunterova bolest, ima različite kliničke uzroke. Hurlerova bolest je najteži oblik, a Scheieova bolest je na kraju kliničke patogeneze. Između ova dva oblika napredovanja postoje prijelazi različitih karakteristika. Najistaknutija karakteristika je oštećena razgradnja ugljikohidrata koji se akumuliraju u lizosomima stanica.
Bolesnici s Hunterovom bolešću mogu imati kratki rast, uvećanu slezenu i jetru, grube karakteristike, zadebljanu kožu, prošireni jezik i otežano disanje. Osim toga, kostur se često mijenja u području zdjelice, kralježnice, kostiju ruku i lubanje. Moguće su pupčane i [[ingvinalne kile].
komplikacije
U većini slučajeva simptomi ili komplikacije pojavljuju se vrlo kasno kod ove bolesti. Zbog toga se dijagnosticira kasno, tako da rano liječenje nije moguće u većini slučajeva. Bez liječenja, razne bolesti i oštećenja unutarnjih organa nastaju kako bolest napreduje.
Posebno su pogođeni bubrezi, jetra i slezina. Na ovu bolest može utjecati i srce, što u najgorem slučaju može dovesti do srčane smrti. Nadalje, dolazi do oštećenja bubrega, a oni koji su pogođeni često pate od bolova u nožnim prstima ili na prstima. Do paralize može doći i ako je mozak oštećen ovom bolešću. Jetra i slezina se mogu povećati i prouzrokovati jake bolove.
Nije neuobičajeno da kosti oboljele osobe budu lomljive i bolne. Liječenje ove bolesti pokazuje se teškim. U mnogim slučajevima životni vijek osobe koja je pogođena značajno se smanjuje. Obično nema posebnih komplikacija tijekom upotrebe lijekova. Međutim, ne može se zajamčiti pozitivan tijek bolesti u svakom slučaju.
Ovdje možete pronaći svoje lijekove
➔ Lijekovi protiv bolovaKada trebate ići liječniku?
Gubitak kose, problemi sa zglobovima i poremećaji organa mogući su znakovi bolesti lizosomalnog skladištenja. Posjeta liječniku preporučuje se ako se simptomi ponavljaju ili se pojavljuju iznenada bez pronalaska razloga. Ako su simptomi povezani s već dijagnosticiranom oštećenjem enzima ili drugom ozbiljnom bolešću, potrebno je konzultirati odgovornog liječnika. Neliječena bolest skladištenja može dovesti do demencije, neplodnosti, neuropatija i drugih komplikacija, od kojih su neke opasne po život. Stoga je potrebno ispitati sve moguće simptome, čak i ako nema određene sumnje.
Simptomi bolesti lizosomalnog skladištenja mogu se pojaviti u fazama ili se razvijaju podmuklo, ali uvijek zahtijevaju pregled i liječenje. Pogođeni ljudi najbolje je razgovarati izravno sa svojim obiteljskim liječnikom ili internistom. Stvarna terapija obično se odvija u specijalističkoj klinici za unutarnje bolesti, iako se fizioterapija ili psihoterapija mogu povezati ovisno o simptomima. Konkretno, terapijske mjere su indicirane zbog često negativnog tijeka bolesti.
Terapija i liječenje
Ovisno o tome koliko je rano postavljena odgovarajuća dijagnoza, ove se nasljedne bolesti mogu vrlo dobro liječiti nadomjesnom enzimom, tako da osobe koje imaju utjecaj imaju daleko manje simptoma, a time i bolju kvalitetu života. Ova zamjenska terapija koristi se prema kliničkoj slici.
Osobe koje pate od Gaucherove bolesti nedostaju "enzim ß-glukocerebrosidaza", koji se proizvodi biotehnološki i infundira se u pacijentovo tijelo. Lizosomi djeluju učinkovito i sposobni su apsorbirati tvari iz neposrednog okruženja. Iz tog razloga umjetno korišteni enzimi modificirani su na takav način da se mogu idealno dostaviti lizosomima.
Makrofagi (fagociti) razgrađuju glukocerebrozide koji su se akumulirali u stanicama. Ova se terapija može usporediti s inzulinskom terapijom za dijabetes melitus, s razlikom što to nije hormon koji nedostaje, nego enzim koji se ne opskrbljuje. Tijelo redovito razgrađuje sve tvari, uključujući isporučeni umjetni enzim.
Zbog ovog redovitog razgradnje tvari, pacijenti moraju redovito podvrgavati ovu infuzijsku terapiju do kraja svog života. Enzim zamjenska terapija ne djeluje simptomatski, već se izravno bori protiv uzroka nasljedne bolesti. Liječnici ovu terapiju nazivaju uzrokom. Principi terapije trebaju se koristiti za sve četiri gore spomenute uobičajene bolesti skladištenja.
Pompe pacijenti se također liječe infuzijskom terapijom. Kod ove bolesti, opskrbljuje se nepostojećim enzimom "kiselina alfa glukozidaza" i pomaže razgraditi glikogen koji se nakupio u mišićnim lizosomima. U bolesnika s bolešću tipa "mukopolisaharidoza tip I", lizosomalni enzim "alfa-iduronidaza" nije prisutan ili nije prisutan u dovoljnim količinama. To je jedna od najrjeđih bolesti skladištenja u kojoj se molekule šećera nakupljaju u organima i tkivima.
Ako je proces normalan, enzim razgrađuje mukopolisaharide. Molekule šećera su dugolančane i sudjeluju u razvoju potpornog i vezivnog tkiva, na primjer kosti, kože, zglobova i hrskavice. Ako je normalan tijek razgradnje poremećen zbog nedostatka enzima, u pojedinim stanicama akumuliraju se patološki glikozaminoglikani (GAG). Buduće mogućnosti terapije usmjerene su na uzimanje tableta.
Izgledi i prognoza
Prognoza za bolest skladištenja je loša. Nađeno je da je uzrok poremećaja zdravlja genetska dispozicija. Zakonski zahtjevi zabranjuju liječnicima i znanstvenicima promjenu ljudske genetike. Iz tog razloga, bolest ostaje doživotno i nema izgleda za oporavak.
Liječnik koji se bavi se koncentrira na liječenje simptoma koji se javljaju. Ako se ne liječi, s vremenom će se povećavati razne žalbe. Koštani sustav je oštećen i nastaju problemi organa. U najgorem slučaju postojat će funkcionalne poteškoće u unutarnjim organima i na kraju neuspjeh aktivnosti organa. To prijeti dotičnoj osobi preranom smrću.
Izazov bolesti leži u dijagnozi. Kod velikog broja bolesnika značajni i snažno uočljivi simptomi pojavljuju se tek kasnije u životu. Kao rezultat toga, genetski poremećaj dugo ostaje neprimjećen, a rano liječenje bolesti je teško. Što se postavi dijagnoza, to je nepovoljniji daljnji tijek. U uznapredovalom stadiju bolesti unutarnji organi ili zglobovi su već ozbiljno oštećeni. Potrebne su kirurške intervencije i ako bolest loše napreduje, samo jedan organ donora može spasiti život oboljele osobe. Stoga je rano liječenje bitno za poboljšanu prognozu.
prevencija
Budući da se radi o prirođenoj genetskoj oštećenju koja sprečava ekspresiju enzima, ovu bolest se ne može liječiti preventivno. Međutim, najnovija dostignuća u genetskom inženjeringu mogla bi pružiti pristup na ovom polju.
kontrola
Uz ovu bolest, ljudi pate od niza različitih komplikacija i bolesti. U pravilu, sve to ima vrlo negativan učinak na kvalitetu života oboljele osobe, tako da dijagnozu treba postaviti vrlo rano. Što se prije savjetuje liječnik, to je bolji daljnji tijek ove bolesti obično.
Ozbiljnost ove bolesti može biti vrlo različita, tako da opće predviđanje često nije moguće. Oni koji su pogođeni pretrpjeli su ozbiljna oštećenja unutarnjih organa. Prvenstveno su pogođeni bubrezi i srce, tako da dijete može umrijeti prvih nekoliko dana ako se simptomi ne korigiraju na vrijeme. Postoje i naslage masti u različitim dijelovima tijela.
Osobito su pogođeni prsti i nožni prsti, što može dovesti do značajno smanjene estetike dotične osobe. U pravilu dolazi do oštećenja bubrega i mozga u daljnjem toku, tako da osoba koja je pogođena umre kao posljedica ove štete. Roditelji i rodbina također često pate od depresije ili drugih mentalnih poremećaja zbog bolesti.
To možete učiniti sami
Bolesti lizosomskog skladištenja vrlo često zahtijevaju intenzivnu medicinsku njegu. Često nema dovoljno mogućnosti za samopomoć. Roditelji pogođene djece često doživljavaju snažan stres u svom kućnom okruženju jer im dijete treba stalnu njegu i pažnju.
Kliničke slike pojedinih bolesti skladištenja su različite. Postoje jednostavni i vrlo teški oblici. Jedan primjer je Gaucherova bolest. Pomoć roditelja često je ograničena na hranjenje djeteta s teškoćama u razvoju. U blažim slučajevima životni vijek može biti gotovo normalan. Bez obzira na to, potreban je stalan medicinski nadzor kako bi se spriječili mogući komplikacije. Redovita tjelesna aktivnost jedna je od popratnih terapija koja se može provoditi i kod kuće. Nadalje, mora se ugovoriti temeljit pregledni pregled raka. To zahtijeva stalne posjete liječniku s djetetom od roditelja. Isto se odnosi i na ostale bolesti skladištenja lizosoma.
U slučaju nekih bolesti, osim tjelesnih oštećenja mogu se pojaviti i mentalna ograničenja koja i dalje zahtijevaju posebnu podršku. U blažim oblicima određenih bolesti, poput Hunterove bolesti, u početku se javljaju samo skeletne promjene i dismorfizmi lica. Ovdje je, međutim, pogođeni pacijent često u stanju voditi neovisan život. Međutim, ovdje su potrebni i stalni liječnički pregledi kako bi se isključile moguće komplikacije poput zatajenja srca ili respiratornih bolesti. Pacijent se može suočiti s psihološkim stresom uzrokovanim fizičkim deformacijama psihološkim savjetovanjem.
.jpg)




.jpg)
.jpg)