kraniodijafizna displazija je prirođena kostna bolest povezana s hiperostozom i sklerozom lubanje lica. Uzrok je genetska mutacija u genima koji inhibiraju koštanu strukturu. Terapija je simptomatska i usredotočena je na zaustavljanje napredovanja bolesti.
Što je kraniodijafizna displazija?

© crevis - stock.adobe.com
Kod hiperostoza koštana tvar se povećava na patološki način. Hiperostoza lubanje je skupina bolesti koja je povezana s takvim povećanjem koštane tvari u području lubanje. Kao kraniodijafizna displazija karakterizira urođena hiperostoza lubanje i koštana je bolest.
Australski liječnik John Halliday prvi je opisao bolest sredinom 20. stoljeća. Učestalost je dana s učestalošću manjom od jednog slučaja u 1.000.000 ljudi. To čini kostnu bolest izuzetno rijetkom displazijom lubanje.
Kompleks hiperostoze i stenoze lica i kranijalnih kostiju seže u genetski uzrok. Zbog nekoliko dosad dokumentovanih slučajeva, nisu sve veze između bolesti konačno razjašnjene. Iz tog razloga, mogućnosti terapije su trenutno ograničene.
uzroci
U velikom broju slučajeva kraniodijafizna displazija se ne javlja sporadično, već s obiteljskom akumulacijom. I autosomno recesivni i autosomno dominantni način nasljeđivanja identificirani su kao način nasljeđivanja bolesti. Autosomno dominantni oblik bolesti zasnovan je na novoj mutaciji gena SOST. Gen se nalazi na lokaciji 17q21.31 i smatra se jednim od najvažnijih inhibitora stvaranja kostiju.
Mutacija SOST gena odgovorna je za veliki broj nasljednih bolesti kostiju, poput VDB. U slučaju mutacije, gen više ne može ispunjavati svoje inhibitorne funkcije i koštana se struktura širi. To u osnovi razlikuje hiperostozu kraniodijafizne displazije od ostalih hiperostoza.
Većina ovih bolesti temelji se na disfunkciji osteoklasta ili osteoblasta. Smatra se da je genetska dispozicija dokazana u vezi s bolešću. Koji još faktori igraju ulogu u nastanku bolesti nije do kraja razjašnjeno.
Simptomi, tegobe i znakovi
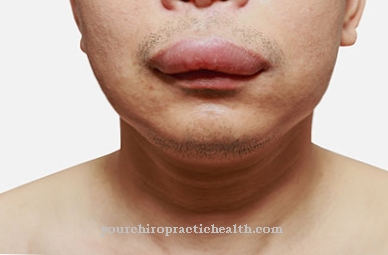
Kliničku sliku kraniodijafizne displazije karakteriziraju različiti klinički kriteriji koji se već očituju u dojenačkoj dobi. Dojena djeca obično imaju jako začepljene nosne prolaze, što im može uzrokovati probleme s disanjem. U kasnijem tijeku bolesti u većini slučajeva postoji potpuna opstrukcija nosnih prolaza.
Često pacijentove suzne kanale nakon ovog fenomena blokiraju. Na donjoj čeljusti većine pogođenih, postupno rastu nosne izbočice koštane tvari. Hiperostoza lubanje lica napreduje i razvija se u leontiasis ossea. U većini slučajeva razvoj zuba pacijenta je poremećen ili odgođen. Unutrašnjost lubanje sužava se kako bolest napreduje.
Suženja također utječu na foraminu i uzrokuju uzastopnu optičku atrofiju. Ovo može biti popraćeno simptomima poput gubitka sluha i manje ili više jake glavobolje. U nekim slučajevima, kako unutrašnjost lubanje postaje sve uska, pacijenti također pate od napadaja. Osovine dugih cjevastih kostiju sve se više proširuju.
Dijagnoza i tijek bolesti
Najranija moguća dijagnoza i naknadna terapija značajno poboljšavaju prognozu bolesnika s kraniodijafiznom displazijom. Vjerojatno liječnik sumnja na hiperostozu vizualnom dijagnozom. Postupci snimanja smatraju se najvažnijim dijagnostičkim alatom. Na primjer, rendgenski snimak pokazuje ekstremnu hiperostozu i sklerozu svih kostiju lubanje.
Ogrlice ili rebra mogu se prikazati proširenim u slikanju. Dijafizi koji nedostaju na dugim kostima jasno se ističu. Delikatni korteks koji se ne može zadebljati također se uklapa u kliničku sliku. U pogledu diferencijalne dijagnoze mora se razlikovati od bolesti poput Engelmannovog sindroma. Molekularno genetske analize posebno su prikladne za takvu diferencijalnu dijagnozu. Engelmannov sindrom pokazuje promjene u genu TGFB1 na mutacijskoj analizi, dok kraniodijafizna displazija utječe na gen SOST.
komplikacije
Kraniodijafizna displazija je rijetka, genetski određena kostna bolest. Simptom se manifestira izravno na lubanji lica kroz snažno povećanje koštane tvari s pratećom sklerozom. Genetska mutacija već je vidljiva u dojenačkoj dobi na temelju oblika lubanje i nepravilno položenih nosnih prolaza, što može uzrokovati prijeteće probleme s disanjem.
Posljedice posljedica kraniodijafizne displazije oboljelom pacijentu donose brojne komplikacije koje su ograničavale život od dojenačke dobi. Ako nema pravovremene kliničke intervencije, rast viška kosti će napredovati. Unutrašnjost lubanje sužava se, a redovi zuba se ne formiraju na odgovarajući način. Zgušnjavajući koštani materijal sužava slušni kanal i postoji rizik od oštećenja sluha, pa čak i gubitka sluha.
Sve više nedostaje prostora u kranijalnoj šupljini, a koštane naslage prodiru u mozak. Jake glavobolje, napadaji, paraliza lica i epilepsija nastaju kao i smanjenje ili regresiranje mentalno stečenih vještina. Roditelji čija su djeca pogođena kraniodijafiznom displazijom trebaju stoga potražiti kliničke mjere u ranoj fazi.
Nakon slikovnog pregleda diferencijalna dijagnoza postavlja se u okviru danih mogućnosti. Trenutno ne postoji osnovna terapija za kraniodijafiznu displaziju. Čine se pokušaji suzbijanja nekontroliranog napretka rasta kostiju i njegovih posljedica. Razni lijekovi kao i dijeta sa smanjenim kalcijem od dojenačke dobi pomoći će oboljelom da smanji simptome.
Kada trebate ići liječniku?
Kraniodijafizna displazija često se dijagnosticira odmah nakon rođenja. U tom slučaju, nadležni liječnik će odmah obavijestiti roditelje, a zatim će izravno započeti liječenje. Ako je displazija manje izražena, dijagnozu postavljaju roditelji. Posjeta liječniku indicirana je ako novorođenče ima poteškoće s disanjem ili vodenastim očima. Vanjske nepravilnosti poput tipičnih malformacija na licu i zubima također ukazuju na bolest koju treba razjasniti i liječiti.
Roditelji koji kod djeteta imaju znakove gubitka sluha ili napadaja trebali bi posjetiti liječnika. Isto se primjenjuje ako se dijete često žali na glavobolju ili daje jaku bol. Tijekom liječenja dijete se mora redovito prezentirati liječniku. To će osigurati da oporavak prođe bez komplikacija. Budući da je kraniodijafizna displazija povezana s nizom simptoma, terapija može trajati mjesecima ili čak godinama. U tu se svrhu liječnik opće prakse savjetuje s drugim specijalistima, uvijek ovisno o simptomima i pritužbama. U liječenje su obično uključeni neurolozi, internisti, specijalisti za uši, kirurzi, fizioterapeuti i psiholozi.
Liječenje i terapija
Uzročna terapija za bolesnike s kraniodijafiznom displazijom još ne postoji. Takva terapija može se zamisliti u budućnosti kroz pristupe genskoj terapiji. Trenutno se, međutim, bolest može liječiti samo simptomatski. Glavni cilj svih terapijskih mjera je zaustavljanje prekomjernog rasta kostiju. Treba poduzeti različite korake.
Napredovanje bolesti, na primjer, može se zaustaviti lijekovima. Kalcitriol i kalcitonin uglavnom se koriste kao lijekovi. Budući da struktura kostiju ovisi o kalcijumu, dijeta sa smanjenim kalcijem može imati smisla. Ovu određenu dijetu treba koristiti dugoročno i idealno pratiti pacijentov čitav život.
Liječenje lijekova bolesnika s umjetnim glukokortikoidnim prednizonom također je pokazalo pozitivne učinke. Što je ranije započela terapija, to je veća perspektiva. Izuzetno ranim liječenjem hiperostoza se može zaustaviti u prvim godinama života. Na taj se način drastično smanjuju sljedeći simptomi.
Pod određenim okolnostima, kao dio terapije mogu se izvršiti i kirurške korekcije. Međutim, takve korekcije obično imaju malo smisla prije nego što je tijek bolesti stavljen pod kontrolu.
Izgledi i prognoza
U kongenitalnoj, ali vrlo rijetkoj kraniodijafiznoj displaziji postoji nepopravljiva genetska mutacija. Stoga prognoza za oboljele nije baš dobra. Medicinski radnici mogu samo pokušati liječiti simptome i posljedice pojačanog rasta kostiju u području glave. Terapija može samo odgoditi tijek bolesti. Kod kraniodijafizne displazije porast koštane tvari je nezaustavljiv.
Budući da današnje mogućnosti terapije ne mogu poništiti temeljnu mutaciju u fazi embrija, druge generacije oboljelih će patiti od nje. Obiteljska akumulacija uočljiva je kod kraniodiafiza displazije. Simptomi povezani s kraniodijafiznom displazijom već se mogu primijetiti kod novorođenčadi. Budući da se sve adhezije kostiju odvijaju u području lubanje, na njih utječu gornji dišni putevi, kao i sluh ili vid.
Osim toga, unutrašnjost lubanje sve je više pod utjecajem stvaranja kostiju. To ograničava terapijske pristupe za naknadne žalbe. Što se ranije dijagnoza može postaviti, to su dugoročne prognoze bolje. Dijeta s niskim udjelom kalcijuma spriječit će povećanje rasta kostiju. Pored toga, odgovarajući lijekovi i prednizon mogu se davati već u ranoj dobi.
Interdisciplinarna strategija liječenja postiže najbolje rezultate. Hirurška intervencija u kraniodijafiznoj displaziji ima smisla samo ako je napredovanje bolesti uspješno obuzdano.
prevencija
Za sada ne postoje preventivne mjere za kraniodijafiznu displaziju. Bolest je genetska bolest koja je povezana s obiteljskim raspoloženjem. Stoga se samo molekularno genetsko savjetovanje može koristiti kao vrsta preventivne mjere.
kontrola
U većini slučajeva dotična osoba ima na raspolaganju vrlo malo mjera za praćenje. U nekim se slučajevima to može čak i potpuno ograničiti, tako da osoba koja je pogođena ovisi o čisto simptomatskom liječenju bolesti. Samoizlječenje se ne može dogoditi jer je to genetska bolest.
Stoga, ako dotična osoba želi imati dijete, treba obaviti genetski pregled i savjete kako se bolest ne bi pojavila kod djece. Sam tretman obično se provodi uz pomoć različitih lijekova koji mogu trajno ublažiti i ograničiti simptome. Uvijek je važno osigurati da se uzima redovno, pri čemu se mora poštivati i ispravna doza.
U slučaju djece, roditelji bi posebno trebali provjeriti jesu li uzeta i pravilno korištena. Redovni pregledi liječnika također su potrebni kako bi se trajno provjerilo stanje bolesti. Većina malformacija može se ispraviti kirurškim zahvatima. Mnogi od oboljelih ovise i o psihološkoj podršci vlastite obitelji u svakodnevnom životu, što pozitivno utječe na daljnji tijek bolesti. U pravilu, ova bolest ne smanjuje životni vijek pacijenta.
To možete učiniti sami
U slučaju kraniodijafizne displazije, pogođeni pacijent ima na raspolaganju samo ograničene učinkovite mjere koje pozitivno utječu na tijek bolesti. Prije svega je odgovarajuća terapija kraniodijafizne displazije od strane tima stručnjaka. Bolest se počinje očitovati u dojenačkoj dobi, tako da su kvalitetu života dotične djece prvenstveno roditelji. U slučaju bilo kakvog bolničkog boravka djetetovih pacijenata, često ima smisla ako su roditelji prisutni u bolnici i dijete kao rezultat toga dobije emocionalnu podršku.
Tijekom bolesti često postoje poremećaji u razvoju zuba, tako da pacijenti često ovise o ortodontskoj terapiji. Potrebna je i vaša vlastita suradnja kada je u pitanju nošenje grudnjaka. Postoje i dokazi da dijeta s niskim udjelom kalcija može suzbiti progresiju kraniodijafizne displazije. I ovdje su pacijenti značajni u slobodi s obzirom na njihovu suradnju, a samim tim i na kvalitetu svog života.
Zbog problema s disanjem pacijenti se odreknu određenih vrsta sporta, ali također vježbaju jačanje kod kuće s fizioterapeutom ako je to medicinski dozvoljeno. Djeca s kraniodijafiznom displazijom dobivaju odgovarajuće obrazovanje u posebnim školama.
.jpg)









.jpg)